
RESEARCH INTRODUCTION
A new way to look for a needle in the ocean
-development of a “digital” protein screening system
Yi Zhang
Accurate high-throughput screening based on digital protein synthesis in a massively parallel femtoliter droplet arrayYi Zhang, Yoshihiro Minagawa, Hiroto Kizoe, Kentaro Miyazaki, Ryota Iino, Hiroshi Ueno, Kazuhito V. Tabata, Yasuhiro Shimane, Hiroyuki Noji
Science Advances, 2019, 5, 8, eaav8185
Japanese English Chinese
High-throughput screening (HTS) for a gene mutagenesis library is like “looking for a needle in the haystack.” While a four-character idiom with the same meaning in Chinese is “looking for a needle in the ocean.” The scientist from SUGAR program of JAMSTEC, Dr. Yi Zhang, and his collaborators developed a general research tool to simplify the way to look for good enzyme mutants in the large protein library.
Advances in science always rely on breakthroughs in technology. In the field of protein engineering, the creation of high-performance proteins (e.g., fluorescent proteins, enzymes, and so forth) heavily relies on the HTS experiment. Existing HTS technologies are time-consuming and expensive because of the low accuracy of hit selection. This study is aimed at improving the accuracy of candidate determination as well as the efficiency of HTS.
- Key Points ■ A new technology to prepare ultra-stable and extremely uniform femtoliter droplets in an ultrahigh density of over one million droplets per 1 cm2 on a thin glass substrate.
- Overview In conventional enzyme screening, the apparent signal intensity associated with the activity of each mutant clone is always masked by a fluctuating protein expression level in cells, causing false-positives or false-negatives in the candidate determination. It is less confident to conclude an activity improvement unless the quantity (or concentration) of every mutant is precisely measured one by one, which is a highly time-consuming and expensive process for a large candidate pool. This work proposed a simple strategy insusceptible to the fluctuation of the protein expression level to enable an accurate determination of the improved mutants, without the need for the measurement of the absolute concentration of every mutant.
■An integrated system to enable a massively parallel protein synthesis reaction in the femtoliter droplet array and to recover single DNA molecules from a single droplet.
■A statistical model with unprecedented accuracy to rapidly identify the improved enzymes from a large mutagenesis library.
Yi Zhang (a scientist of SUGAR Program, X-star, Japan Agency for Marine-Earth Science and Technology), in collaboration with scientists from UTokyo, AIST, and IMS, established a method to prepare a high-density femtoliter droplet array (FemDA) consisting of 1 million uniform, stable, and biocompatible droplets per 1 cm2 (Fig. 1). The droplet encapsulated DNA and all components necessary for cell-free protein synthesis. In particular, the DNA molecules were randomly distributed into each droplet following a Poisson process, which allows 0, 1, 2, 3, ……DNA molecules to be encapsulated in the droplets. As demonstrated with many kinds of proteins, the protein yield in a droplet was nicely proportional to the number of encapsulated DNA molecules (Fig. 2). Here, the fluorescence intensity reflected the relative protein quantity. Thus, the maximum protein yield (relative) in a single droplet can be predicted using Poisson statistics, as the total number of droplets and the DNA concentration in a given experiment are generally known. In other words, the physical number of DNA molecules in a single droplet must be under the restriction of Poisson distribution (Fig. 3). If there are some droplets showing signal intensities higher than the Poisson-predicted maximum in a given array, such droplets should contain the improved mutant rather than containing multiple DNA molecules (i.e., higher protein yield). Because the discrete distribution of signal intensity is the basis of setting the unambiguous selection threshold, Zhang named this method “digital screening” or “digital HTS.” This is a new digitalized biotechnology after digital polymerase chain reaction (digital PCR) and digital enzyme-linked immunosorbent assay (digital ELISA) (Fig. 4).
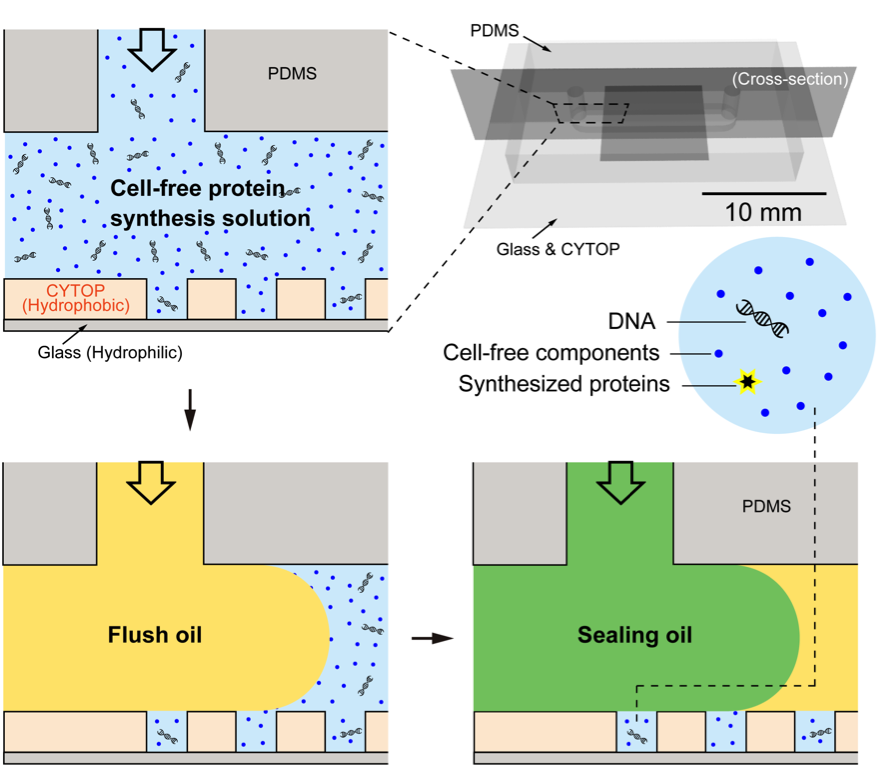
Fig. 1. Schematic illustration of the preparation of femtoliter droplet array (FemDA) used for cell-free protein synthesis (CFPS) driven by single DNA molecules.
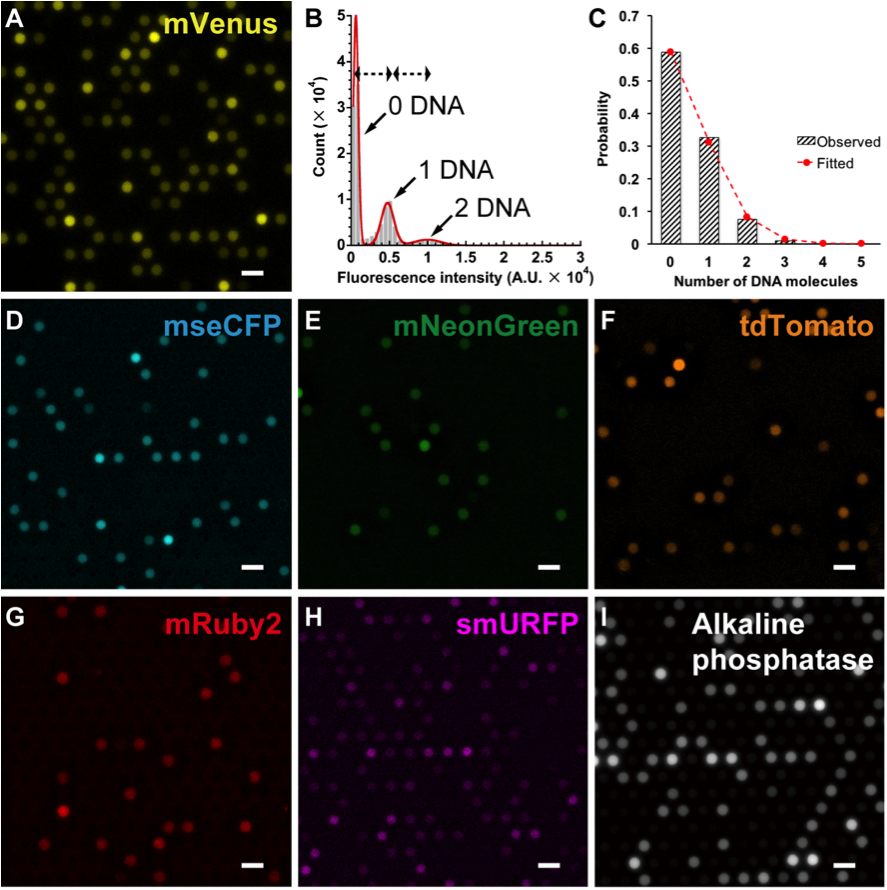
Fig. 2. CFPS from single DNA molecules with FemDA. (A) mVenus fluorescent protein synthesis. (B) Histogram of fluorescence intensities of droplets from an array. The histogram showed a discrete distribution of fluorescence intensities corresponding to the quantity of proteins in every droplet. The histogram was well fitted by a sum of Gaussian distributions of the equal peak-to-peak intervals, which suggests an occupancy of different numbers of DNA molecules per droplet. (C) Histogram of the DNA occupancy in a droplet. The probability of occurrence of droplets containing different numbers of DNA molecules up to 5 (the largest number observed in the experiment of Fig. 2A) was perfectly fitted by a Poisson distribution, as expected for a random distribution of DNA molecules. (D-I) Several other examples of CFPS for fluorescent proteins and an enzyme. Scale bars,10 μm.
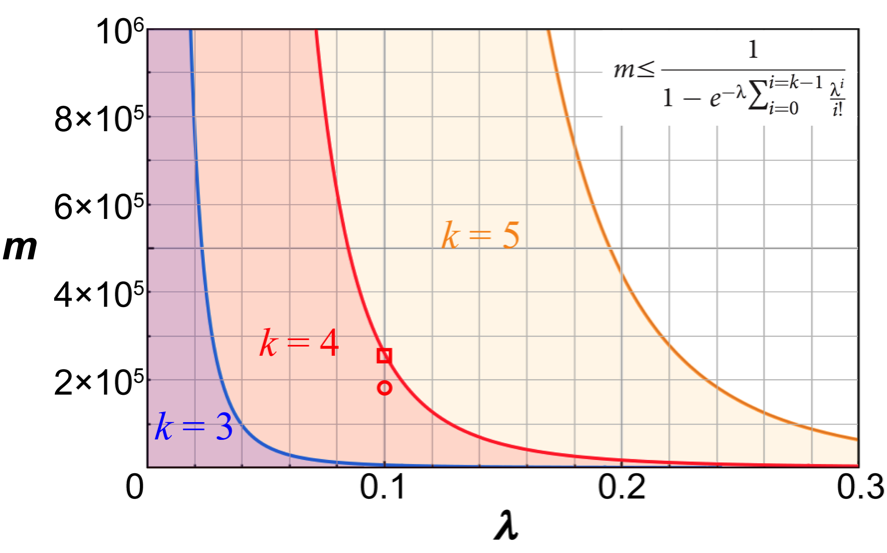
Fig. 3. The mathematical model of digital screening. The inequality in the graph shows the relationship between the total number of droplets (m) and the DNA concentration (λ, the average number of DNA molecules per droplet). Parameter k represents the maximum physical number of DNA molecules in a droplet. It is clear that λ and m are preferably less than a threshold (the semitransparent color-shaded area), respectively, to avoid false positives. The throughput (the number of droplets) of a screening experiment is not simply “the more, the better.”
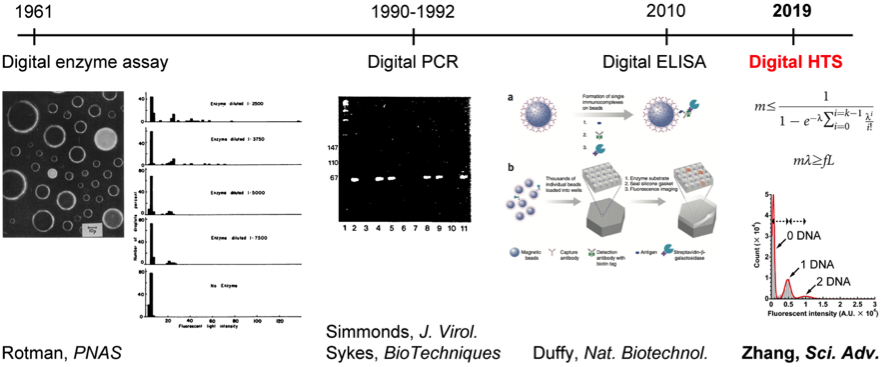
Fig. 4. The generations of digitalized biotechnologies. The digital HTS led the biomolecules from the detection to the creation.
The first trial screened a single-site saturation (each amino acid residue is randomly substituted by one of 20 natural amino acids) mutagenesis library of Escherichia coli alkaline phosphatase covering its 48 amino acids. The DNA library was introduced into 180,000 droplets at a concentration of 0.1 DNA molecules per droplet on average (the circle coordinate in Fig. 3), so the total number of droplets containing 4 or more DNA molecules should be less than one. There were a few droplets with the fluorescence intensity higher than the theoretical maximum on the chip (Fig. 5). These femtoliter droplets were individually recovered by a microcapillary (Fig. 6). The DNA in each droplet was amplified by PCR and subjected to DNA sequencing. Surprisingly, these mutants not only included a mutant that has been confirmed as the most active one in the past decades but also two new mutants that have never been discovered and even never been taken into consideration, despite the fact that this enzyme was well characterized and extensively studied. This experiment initially designed for a positive control to see whether the aforementioned known mutant can be rapidly identified from a large library killed two birds with one stone. How little about the protein structure-function relationship we know!
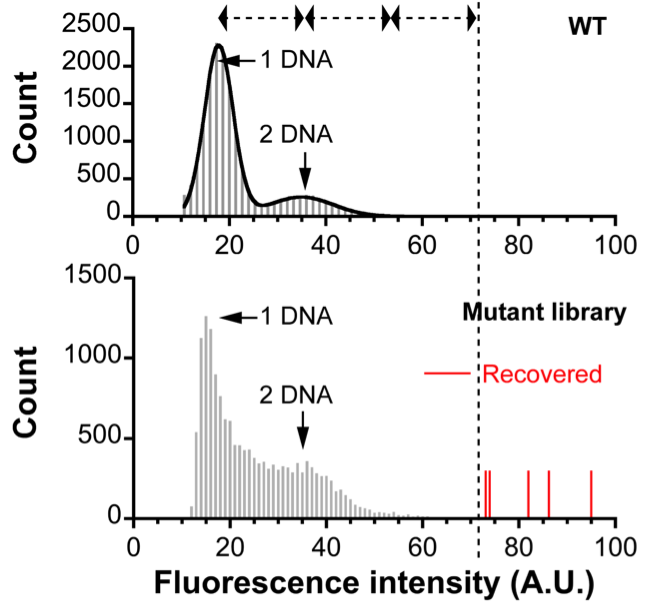
Fig. 5. Digital screening of an alkaline phosphatase library. A few droplets (red label) showed fluorescence intensities higher than the theoretical maximum (vertical dotted line). WT, wild-type.
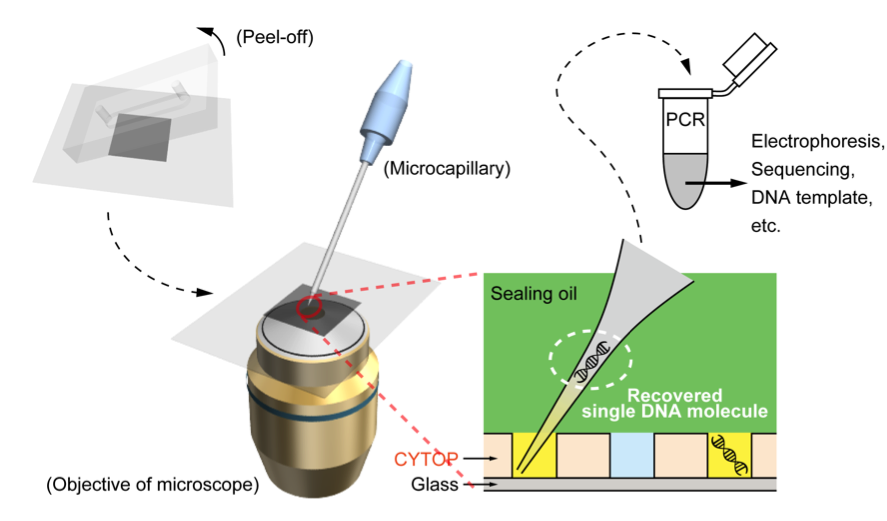
Fig. 6. Single-molecule DNA recovery and amplification.
As suggested by the mathematical model of digital screening, there must be a boundary condition (solid line in Fig. 3) in the setup of experimental conditions. Approaching the boundary condition allows more droplets, as well as higher throughput, can be applied to cover a larger library (the screening throughput corresponding to the square coordinate of Fig. 3 is higher than that of the circle coordinate). Based on this experimental setting, this study also attempted another digital screening for a relatively new alkaline phosphatase cloned from a psychrophilic marine bacterium, Cobetia marina. The wild-type has been recognized as the most active alkaline phosphatase among known microbial or mammalian ones. The screening rapidly identified two mutants with the same mutation site, which was substituted by either lysine or arginine, the only two strongly positively charged amino acids among the 20 natural amino acids. The activity improvement was specific toward the fluorinated substrate used in the screening, suggesting a reinforced electrostatic interaction between the unnatural substrate and the positively charged residue. This screening was carried out within a limited time at the request of a reviewer, who placed an order of an additional target other than the Escherichia coli alkaline phosphatase. The success reinforced the proposed digital screening strategy and suggested that a highly active enzyme may be further evolved toward unnatural substrate because of the potential lack of selective pressure in nature. How interesting the one-day molecular evolution is!
Because of the almost infinite number of combinations of mutation sites in a protein, the libraries we can handle are just a tiny portion of all possibilities. It is still highly possible that a library contains no good mutants. The digital screening scheme can quickly (within hours!) conclude a useless library just after the high-throughput protein synthesis reaction. This feature is also beneficial for accelerating a screening project through rapidly changing the strategy of library design/preparation, rather than wasting a lot of money and time on repeatedly testing a useless library.
With the advances in bioinformatics and library construction techniques, the era of a prompt on-demand creation of high-performance proteins will be surely coming.
Back to top